A Slice of Biology - A Review on Confocal Microscopy
Introduction:
The use of microscopes has always been critical for research in Biology and in the last 100 years, microscopy has come an extremely long way. Many new technologies have been developed and one of the most influential is Confocal Microscopy. Confocal microscopy has existed for more than 60 years, however, has really only become practical in the last 30 with advances in technology. Marvin Minsky, during his time as a post-doctoral fellow at Harvard in 1955, created the first variation of the confocal microscope.^(1,6,7) He did this to try and observe neural networks in living brain tissue, however, the technology and computing power at this time were not advanced enough to allow this technology to be fully functional. As will be seen in later sections of this paper, confocal microscopy heavily relies on computational power to not only control the creation of the images but also to compile the data collected. Without computer power, Minsky had to use mechanical means to build the images, which resulted in a lot of distortion and blurring. These blurred images are what caused confocal microscopy to be widely underutilized by the biological research community until the 1980’s when Brad Amos and John White took Minsky’s designs and added two crucial technologies – computational laser scanning, and fluorescence. Their goal in redesigning this microscope was to image specifically tagged parts in the focal plane of dividing cells.^(1,6) With the addition of computational technology to the confocal microscope, images were able to be produced at higher resolution, making confocal microscopy increasingly more popular throughout the biological research community. Today, the technology can be seen in a large percentage of research papers and has become an extremely powerful tool.^(1,6,7)
The benefits and drawbacks of Confocal microscopy will be discussed in depth later however to give a sense of its power: a confocal microscope can produce 0.5-1.5 micrometer optical sections of a specimen while simultaneously having higher resolutions than wide field microscopes.^(1) Since optical sections of a specimen are created, if multiple sections at slightly different points in the specimen are stacked together, a 3-dimensionally reconstruction can be generated. This high degree of resolution and optical sectioning is obtained through precise optics that will be explained in more detail in the “Physical Mechanism” section. The only real drawback to confocal microscopy, is that only a few different excitation wavelengths can be used.^(1,8) With these major benefits and only minor drawbacks, it is clear why confocal microscopy has become so prevalent in the biological research community.
Physical Mechanism:
The parts behind a confocal microscope are crucial to the microscope's ability to bend light in the way it does. For ease in explaining the mechanism of this microscope, fluorescence confocal microscopy will be described initially, then related back to other forms of confocal microscopy. The parts of a fluorescence confocal microscope include a laser which provides the excitation light, a dichroic mirror, galvanometer scanning mirrors (not shown in figure), an objective lens and a detector aperture. The laser provides parallel rays of light for the excitation source that allow the optical sections to be obtained. The dichroic mirror is a surface that reflects most wavelengths of light except a very specific range that, instead of being reflected, will readily pass through. The galvanometer scanning mirrors are small surfaces that are controlled by motors to reflect the laser light such that the light can move (scan) across the specimen. Lastly, the detector aperture is a small pinhole disk that can change in size in order to let different amounts of light through.^(1,2) These four pieces, as will be explained, are crucial for the function and versatility of the confocal microscope.
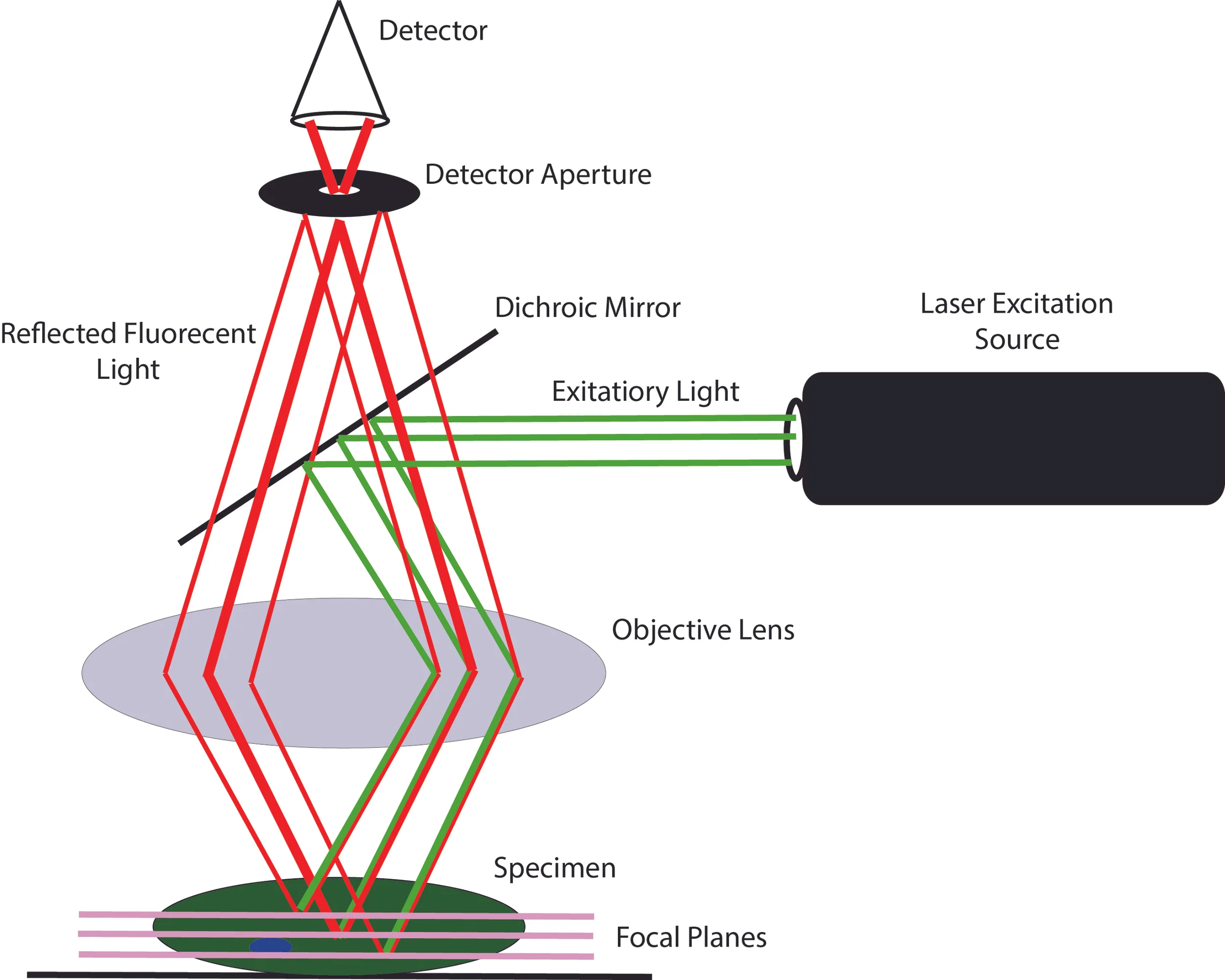
As for the setup of the fluorescent confocal microscope, there are two slightly different configurations that can be utilized.^(2) The first configuration, and the most widely used, simply shoots parallel rays of light at the dichroic mirror which will reflect off and into the objective lens. The objective lens will then refract, or bend, the light onto the specimen. This refraction will create different “planes” of light stacked on top of each other in the specimen. This allows the focus to be adjusted on the microscope such that only one of the focal planes created in the specimen will be picked up by the detector. This is done by only allowing one focal plane’s light to perfectly reflect off the specimen, refract through the objective lens, and go into the detector. The key thing about the light coming from the specimen is that, since it is interacting with fluorescent molecules in the cell, the light loses energy and thus has a slightly longer wavelength than the excitation light and can readily pass through the dichroic mirror. The other key part of this setup is the detector aperture which acts to block all of the light rays not in the plane being focused on. Light from anywhere other than the plane of focus will not be reflected and refracted at the correct angle to go through the aperture and into the detector, thus, all out of focus light is eliminated and an optical section of the specimen is created.^(1,2,4) To make this system even more interesting and complex, the laser light is a single point, and does not cover the whole specimen. This means that the light has to be scanned across the entire specimen to create an image of the plane that is desired to look at. This scanning is done by the coordinated movement of the galvanometer scanning mirrors to effectively move the beam of light across the specimen. Originally, instead of moving the laser beam, Minsky achieved this scanning technique by moving the stand (with the specimen on it) under the light to gather data on the entire plane. As stated before, this caused high amounts of distortion due to minute movements of the specimen on the slide and movement of the stand in the z-axis that was not intended.^(1) This was one of the main reasons confocal microscopy was not applicable until almost 30 years after its conception, since the computational power was physically not available yet to scan the beam of light like is done today. With all of this being said, if the optics of this system are not perfect, some of the light from other planes may come into the detector and create blurs in the field of view, however, this creates much less distortion than moving the entire specimen like Minsky had done.^(1,2,4)
Occasionally with confocal microscopes, since everything is so precise, there is a problem of not having perfectly parallel laser light to get clear images. This is where the second configuration of the fluorescent confocal microscope comes in since this setup can fix the quality of laser light. This is done by having the laser light be sent through a pinhole aperture (like that at the detector) that will cause only parallel light to pass through. Since light acts as a wave however, when it passes through this pinhole aperture, it will diffract, or spread out evenly in all directions. Even though this diffraction occurs, as the light is reflected off of the dichroic mirror and into the objective lens, the light rays will become parallel again and act just as they would in the first configuration.^(1,2) Both of these configurations accomplish the desired outcome, however, using the first configuration is much more ideal. With the aperture in front of the excitation source, a significantly less amount of light is able to be shone on the specimen since only a fraction of the excitation light is getting through the aperture.^(2,6) This causes a need to have light shone on the specimen for longer than normal and since not as much light is being shone on the specimen, not as much light can be detected and there is a possibility the confocal microscope will produce lower quality images.
While the precision and functionality of the mechanisms in a confocal microscope are crucial, the compiling of collected data and controlling of mirrors is just as, if not more, important. This is where the scan head is important. The scan head is a mini computer that is responsible for collecting and controlling all of the position and optimization data from the different mirrors and lenses of the microscope. Along with this, the scan head is also responsible for combining all of the photon data collected to create one image. When working properly, this system will capture and compiled all the data needed into one image in about 1 frame (focal plane) per second.^(1,8) To achieve faster rates of capture than this, a system much more complex than the one described is required in which multiple beams of light scan the same specimen.^(1)
Applications:
One of the most widely used applications of Confocal Microscopy is its imaging of fluorescence. Fluorescence is the idea that a molecule, with a fluorescent domain (that can be excited by a light source) and a binding domain for a specific protein, is introduced into a cell and allows the location of that protein to be visualized. Since such a high energy light source is being used in a confocal microscope, it is logical that almost from its beginning, confocal microscopy has been paired with fluorescence. This is not to say confocal microscopy can’t be used for other techniques though. Confocal microscopy can be used without fluorescence in a technique called reflectance confocal microscopy which uses the same principles as fluorescence, however, the light takes a more complex pathway.^(7) The reason for this is that a dichroic mirror cannot be used since the excitation light does not interact with any fluorescent molecules in the cell and thus does not change wavelength. Though the basic principle of sectioning the specimen in fluorescence confocal microscopy is the same in reflectance, the entire setup of the microscope is different due to the fact that dichroic mirrors cannot be used.^(7) Another form of confocal microscopy that is even more complex than either of those previously discussed is one called Resonant Scanning Laser Confocal Microscopy. This form of confocal microscopy involves the use of two or more excitation sources that scan across a specimen at the same time in order to drastically increase the speed of image formation. In a fluorescent confocal microscope, the image capture rate is about one frame per second, however, in resonant scanning confocal microscopy, the frame rate jumps to thirty or more frames per second.^(9) This being said, as the frame rate gets higher than thirty frames per second in the resonant scanning microscope, the resolution at which the images can be formed progressively decreases due to how fast the lasers are having to scan.
These forms of confocal microscopy are only some of the variations, however, as can be seen, the ability of confocal microscopes to image sections of a cell can be extremely powerful in multiple different contexts. To make it even more powerful, it is possible to use the images that are created to construct a 3-dimensional model of a specimen. This can be done by taking tens of images of a specimen, each with a slightly different focus and thus each image being a slightly higher section of the specimen. These images can then be combined using computer programs in order to reconstruct a 3-dimensional image of the specimen.^(1) This method can also incorporate fluorescence, allowing you to view the location of fluorescence in a 3-dimensional rendering of the specimen.^(1) This combination of techniques can be especially useful when looking at complex structures such as the cytoskeleton of a cell or even a network of interconnecting neurons.
Amazingly, the enhancement of confocal images does not stop there. If a zoomed-in section of a specimen is desired, instead of changing the objective lens to create a greater magnification, the microscope can gather data on a smaller section of the specimen. This means the laser would do its normal scanning pattern except on a smaller region of the specimen, creating a zoomed in image at the exact same resolution as an image of the full specimen.^(1,7) This process is called Zoom Factor.^(1)
Uses in Research:
As has been outlined above, confocal microscopy can be extremely useful in research. One very intriguing example of this uses 3D Confocal Microscopy to analyze the different roles of two different cell types - Astrocytes and Brain Microvascular Endothelial Cells - in the animal form of Multiple Sclerosis (MS). Both of these cell types are known to express a receptor called CCL2 that becomes extremely up-regulated and drives neuroinflammation in MS. What is not know however, is whether there is any difference in how each of these cell types contributes to the disease. In order to figure this out, Debayon Paul and Shujun Ge, et. at. conducted research that used two different mouse strains, one with the astrocyte version of CCL2 knocked-out (KOed) and the other with the endothelial version of CCL2 knocked-out. This allowed Paul and Ge et. al. to observe how the disease progresses with one of the two cell types not functioning to affect the disease. To observe the progression of the disease in each case, the test mice were sacrificed and their spinal cords were removed and frozen in cryomatrix. After freezing, the researchers sectioned the spinal cords, double stained them with antibodies for the molecules being looked at, and then fixed the sections to a slide. These slides were then imaged using confocal microscopy to assess the inflammatory response in the CNS and see if and how Leukocytes were able to penetrate the blood brain barrier (BBB), indicating progression of MS. From these experiments it was found that with CCL2 KOed in the astrocytes there was a complete lack of BBB penetration by the Leukocytes. In the endothelial cells with CCL2 KOed, however, they found Leukocytes stalled in the microvascular lumen, signifying a partial start to crossing the BBB.^(1)0 Without the use of confocal microscopy it would have been extremely difficult to get as strong of results as Paul and Ge et. al. did because the position of the Leukocytes would have been hard to distinguish from the rest of the sample.
Confocal microscopy has proven to be an extremely diverse tool for research and that is shown again with a recent paper describing its use in diagnosing different kinds of skin cancer. The study done by Guitera, et. al. used confocal microscopy to identify 47 different features of skin cancer cells (some of which had already been described) and calculated their frequency of appearance among the different lines of skin cancer used. Using these frequencies, they were successfully able to identify which cancer each sample was with slightly more accuracy than the conventional histopathology tests.^(1)1 There was not a significant difference between the identification using confocal microscopy or histopathology tests, however, the use of confocal microscopy provides some exciting possibilities. The researchers were not only able to identify the cancers at the same accuracy as conventional methods, but they were also able to identify new features of the cancer cells and calculate their frequency of appearance. From this study, confocal microscopy was shown to be a viable method of cancer identification and may be able to be used in conjunction with typical identification methods. This again illustrates the amazing diversity of confocal microscopy and opens the door for yet another field of study to utilize it.
Discussion & Conclusion:
Over the past 30 years, confocal microscopy has proven to be an exceptionally diverse and useful tool in biological studies. Whether it is used for sectioning of a certain specimen, creating a 3D reconstruction of a cell, or obtaining a better resolution with less background noise, it allows for multiple new doors to be opened in research. With the further enhancement of technology and the continued widespread use of confocal microscopy, it is almost a guarantee that new and innovative uses for this technology will be invented. The diversity and uniqueness of applications that have already been presented has rightfully made it one of the most popular tools for biological research in the last few decades.
Works Cited:
1. Fellers, Thomas J., and Michael W. Davidson. "Introduction to Confocal Microscopy." Olympus Microscopy Resource Center. Olympus America, 17 Dec. 2012. Web. 29 Feb. 2016.
2. Borlinghaus, Rolf T. “Confocal Microscopy.” Leica Sciencelab. Leica Microsystems, 9 May 2011. Web. 19 Mar. 2016.
3. Nanarezai. "Confocal Microscopy - A Visual Slice of the Cellular World." SCQ. N.p., Aug. 2004. Web. 29 Feb. 2016.
4. Lit Guwahati, and B.R. Boruah, Applications of Confocal Fluorescence Microscopy in Biological Sciences. India Institute of Technology. Web.
5. Rigby, Paul J., and Roy G. Goldie. “Confocal microscopy in biomedical research.” Croatian medical journal 40 (1999): 346-352
6. Paddock, Steve. “Over the rainbow: 25 years of confocal imaging.” Biotechniques 44.5 (2008): 643.
7. Paddock, Stephen W. "Basic Concepts." Nikon MicroscopyU | Confocal Microscopy | Basic Concepts. N.p., n.d. Web. 22 Mar. 2016.
8. "Confocal Microscopy - The Comprehensive Explanation." All Microscope. Web. 22 Mar. 2016.
9. Larson, Jeffrey M., Stanley A. Schwartz, and Michael W. Davidson. "Resonant Scanning in Laser Confocal Microscopy." Confocal Microscopy. Nikon MicroscopyU, Web. 22 Mar. 2016
10. Paul, Debayon, et al. "Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation." Journal of neuroinflammation 11.1 (2014): 10.
11. Guitera, Pascale, et al. "In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: analysis of 710 consecutive clinically equivocal cases." Journal of investigative dermatology 132.10 (2012): 2386-2394.
12. Urszula, Pankiewicz, et al. "Visualization of calcium and zinc ions in Saccharomyces cerevisiae cells treated with PEFs (pulse electric fields) by laser confocal microscopy." Food chemistry 188 (2015): 16-23.